This web page was produced as an assignment for Gen677 at UW-Madison Spring 2009.
NrCAM protein domains
The schematic below shows the predicted protein domains of the human NrCAM isoforms A, B and C as well as a number of homologues . The Ig and FNIII domains are important to note as they indicate that the predicted functions and interactions of the NrCAM protein in brain and axonal development are likely. These domains are seen in various proteins with similar predicted/known functions suggesting that NrCAM does in fact play a role in the development of neural networks and the central nervous system. PFAM and SMART returned the same protein domains. PROSITE did not have the same information and did not return any interesting protein domains. The schematic below is modified from the PFAM website.
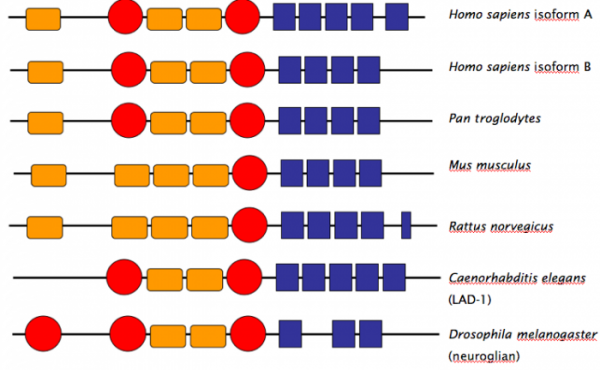

Protein domain analysis
The similarity of the protein structures throughout these organisms will allow researchers to target specific domains in the NrCAM protein in order to find the functionally relevant parts of the protein. The high homology between the human protein structure and the chimpanzee protein structure will be very important in the future as scientists will need to characterize the behavioral effects of NrCAM mutations as autism is very much a behavioral disorder. Cognitive and social deficits will be hard to model in the mouse or worm so it will be important to study the social implications of autism in an organism where behavioral traits can be measured.
The functionality of the protein domains is also worth talking about as the three domains present in the human protein and all the homologues are associated with many of the predicted functions of NrCAM. The I-set domain is involved in cell-cell recognition, cell-surface receptors, and is seen in a variety of other adhesion molecules including vascular adhesion molecules and intercellular adhesion molecules. The Ig domain has been shown to have similar functionality as well as functions in the immune system and muscle structure. The FN3 domain is a glycoprotein domain involved in DNA binding, heparin binding, and cell surface binding as well as cytoskeletal maintenance, cell differentiation, and cell migration functions. These domains support the gene ontology data for NrCAM and provide reason to study the role of NrCAM in autism. In addition, the domains support the proposed localization patterns of the NrCAM protein to the plasma membrane. The functions of the identified domains are all related to the cell membrane.
ClustalW protein alignment
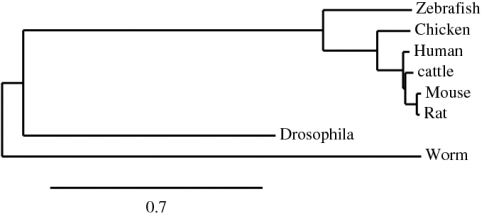
The ClustalW alignment of the human NrCAM protein to the homologous proteins in zebrafish, chicken, cattle, mouse, rat, drosophila, and worms. All default settings for alignment were used [3]. Alignments were done for the four matrices available in ClustalW (Blosum, Pam, Gonnet, Id) producing identical trees. The phylogenetic tree below is a result of the ClustalW alignment using the Blosum matrix.
| clustalw_proteinalignment.pdf | |
| File Size: | 77 kb |
| File Type: | |
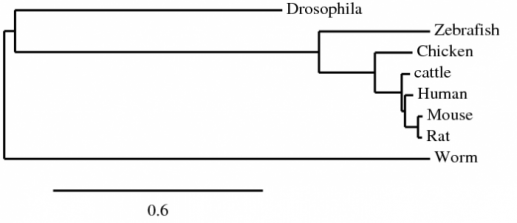
Muscle protein alignment
The Muscle alignment of the human NrCAM protein to the homologous proteins in zebrafish, cattle, mouse, rat, drosophila, and worms. All default settings for alignment were used [1,2]. The phylogenetic tree below is a result of the Muscle alignment.
| muscle_proteinalignment.pdf | |
| File Size: | 92 kb |
| File Type: | |
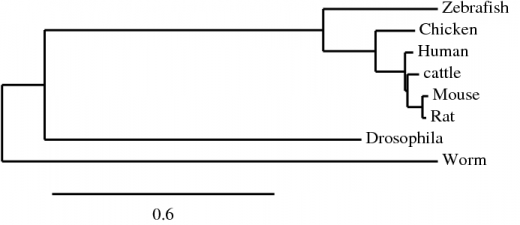
T-COFFEE protein alignment
The T-COFFEE alignment of the human NrCAM protein to the homologous proteins in zebrafish, cattle, mouse, rat, drosophila, and worms. All default settings for alignment were used [4,5]. Alignments were done using both the Blosum and Pam matrices in T-COFFEE producing identical trees. The phylogenetic tree below is a result of the T-Coffee alignment using the Blosum matrix.
| t-coffee_proteinalignment.pdf | |
| File Size: | 77 kb |
| File Type: | |
Paragraph.
Analysis
The three programs used to analyze the human NrCAM protein and its homologues are quite similar in format and ease of use. These programs can all be found at EMBL-EBI and require a FASTA input format. Multiple sequences can be analyzed simultaneously, however the length of the sequence that can be entered is limited. This did not affect my analysis as the human NrCAM protein is within the range. ClustalW seems to be the more useful the Muscle and T-COFFEE based on more matrices for analysis and the ability to change several default settings to optimize results [1,2,3,4,5]. The alignments returned by all three programs showed high protein sequence homology in all sequences with the exception of C. elegans. This would be expected of the mammals in the alignment, but it is kind of surprising that the chicken and the zebrafish are so similar to the mammals. The most variation occurs in the terminal regions of the protein sequences, the rest of the amino acid sequence is pretty similar throughout. All three programs produced phylogenetic trees showing fairly close relation between mammals, and more distant relation between mammals and worms as expected. The position of drosophila is a bit different in each tree as well as the relation between chickens, humans, and cattle with mouse and rat sequences. Overall, I found all three sequence alignment programs to be very easy to use. The programs did not return a lot of excess information that can be found in other databases, instead the results were simple and relatively easy to interpret. The different options for the format of the results was useful as well.
1. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008 Jul 1;36 (Web Server issue): W465-9. Epub 2008 Apr 19. (PubMed)
2. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high thoughput. Nucleic Acids Res. 2004, Mar 19;32(5): 1792-7. (PubMed)
3. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 200, Apr;17(4): 540-52. (PubMed)
4. Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, Oct;52(5):696-704. (PubMed)
5. Anisimova M., Gascuel O. Approximate likelihood ratio test for branches: A fast, accurate and powerful alternative. Syst BIol. 2006, Aug;55(4):539-52. (PubMed)
6. Chevenet F., Brun C., Banlus AL., Jacq B., Chisten R. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006, Oct 10;7:439. (PubMed)
Brett Maricque
[email protected]
Last updated: 5/13/2009
http://www.gen677.weebly.com
